
Please complete the Pre-VTP Survey.
The primary goal of this Virtual Training Project (VTP) is to use the Linux and R programming languages to bioinformatically characterize, quantify and visualize the species composition of urban microbiome samples (i.e. subway swabs) from raw data to completed analysis.
What is bioinformatics
Why is the metagenomics application of bioinformatics useful?
In case you are thinking, "How would someone be able to use this knowledge in real life?", here are three real-world applications of the metagenomics analysis technique you will learn:
Research Applications: you can ask questions about the similarity
of microbiome samples, the prevalence/emergence of antimicrobial
resistant strains, etc. The MetaSUB Project, which is where the data
you'll analyze in this VTP comes from, would be an example of this
application.
Clinical Applications: This technique is already being employed by
some biotech startups, like Karius, to
rapidly diagnose blood-borne infections.
Biotech/Industry Applications: Some companies offer microbial
surveillance services to identify and monitor the presence of
resistant pathogens (e.g. on hospital surfaces). One such startup
called Biotia was spun out of the Mason
lab (the same Cornell Med lab that created the MetaSUB project) and
utilizes many of the same techniques that you will employ in this
VTP."
In a broader sense, bioinformatics is a subdivision of data science; principles learned in the former are highly relevant to the latter. We'll point these out as we go along.
The source of the data you will analyze in this VTP is from the MetaSUB Project, an effort to characterize the "built environment" microbiomes of mass transit systems around the world, headed by Dr. Chris Mason's Lab at Weill Cornell Medical Center.
MetaSUB an international consortium of laboratories created to establish a world-wide "DNA map" of microbiomes in mass transit systems. It is succeeded a previous study
MetaSUB consists of a group of labs from all over the world working together to study the microbes in mass transit systems like bacteria, viruses, and archaea (i.e. their microbiomes) and make a "DNA map" of sequences from them. The project was started by the Mason lab and is an extension of a previous study called Pathomap. Pathomap only looked at the NYC subway system and aimed to create a profile of what's normal, identify potential bio-threats, and use the data to make the city better for people's health. MetaSUB recognized that other cities around the world could benefit from a similar study and decided to expand the project to include subway systems worldwide.
Take a look at this figure, which provides an overview of MetaSUB's design and execution:
As you can see, the researchers collected samples from
(A) New York City's five boroughs
(B) Collected samples from the 466 subway stations of NYC across the 24 subway lines
(C) extracted, sequenced, QC'd and analyzed DNA
(D) Mapped the distribution of taxa identified from the entire pooled dataset, and
(E) presented geospatial analysis of a highly prominent genus, Pseudomonas
Notably, as seen in (D), nearly half of the DNA sequenced (48%) did not match any known organism, underscoring the vast wealth of likely unknown organisms that surround passengers every day.
Now, let's focus on Fig 1C, this is shows at a high level the steps to process and analyze samples. We'll be doing a very similar analysis in this VTP but not identical.
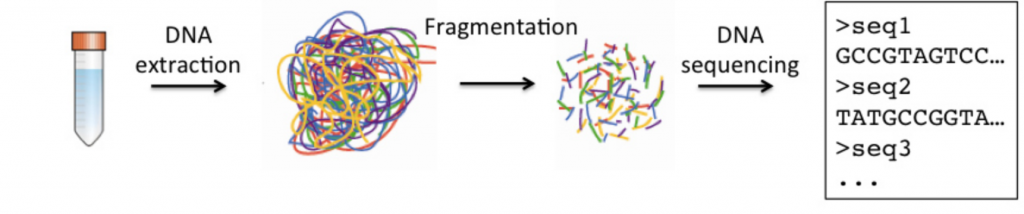
Here's a simplified version of sample processing and sequencing:
Alright, before moving onto the next section, here are some more resources in case you want to review them later:
Here's the MetaSUB paper in case you would like to review it later. To be frank, this article is a bit challenging to read, so we suggest you review it later on if you're inclined, after you've done a bit of the VTP.
New York Times article about the MetaSUB Project.
Here's the Pathomap paper, the predecessor study that only investigated the NYC Subway Microbiome.
Throughout this VTP, you will characterize, quantify and visualize microbial metagenomics data from sequenced swabs of public urban environments on your own cloud-hosted High Performance Compute Instance.
In the Linux terminal, you will learn the structure of raw genomic data and perform taxonomic characterization & abundance quantification. In R, you will determine the mose abundant species in your sample and visualize results at several taxonomic levels.
Here's a flowchart of the analysis you'll perform:
All bioinformatics tasks will be performed in "the cloud" on your own Amazon Web Services (AWS) hosted high performance compute instance.
What is cloud computing:
For this project, you will execute all Linux and R tasks in RStudio.
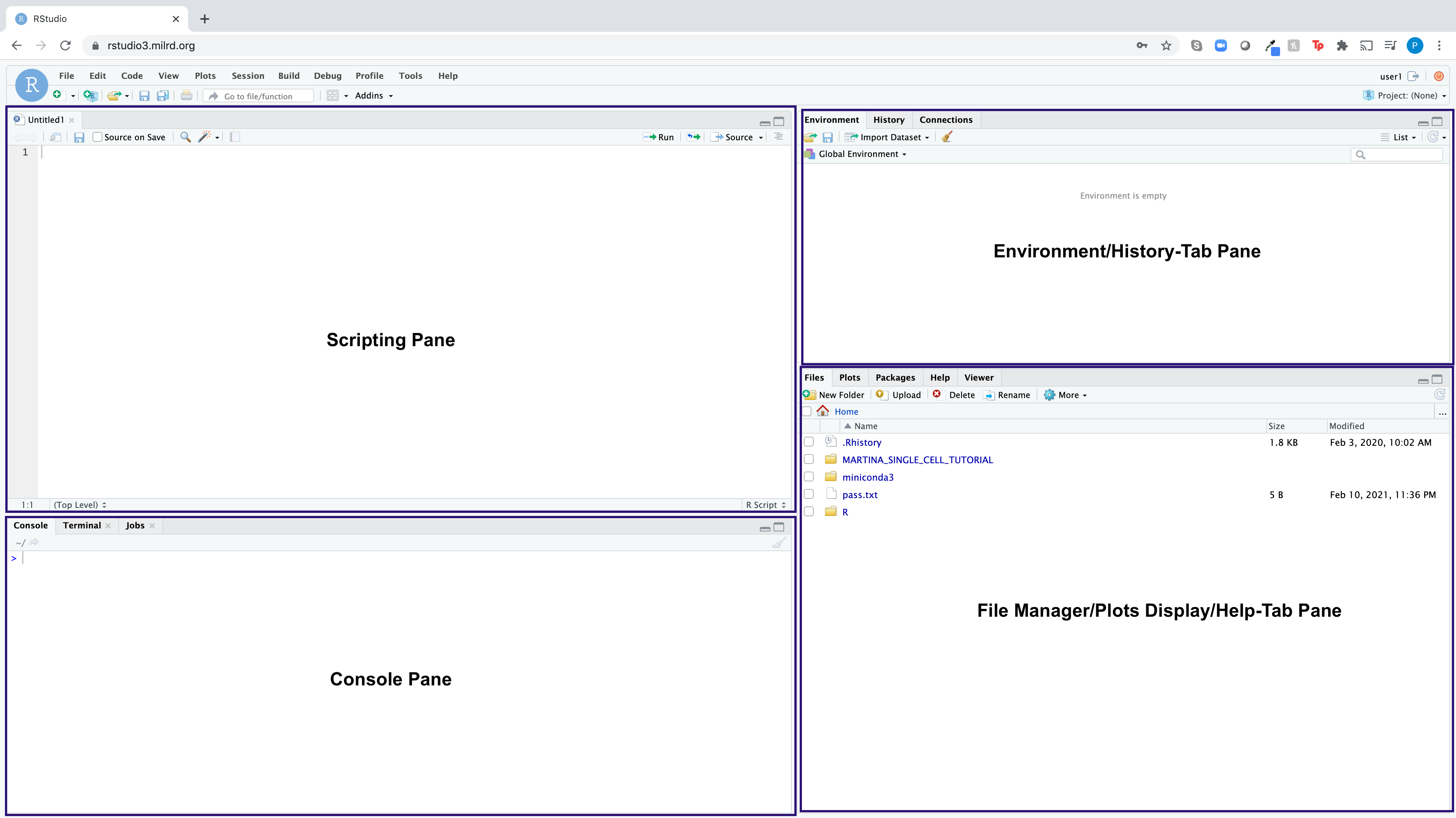
RStudio is an integrated development environment (IDE) for the open-source R programming language, which basically means it brings the core components of R (Scripting Pane, Console, Environment Pane, File Manager/Plots/Help Pane) into a quadrant-based user interface for efficient use.
RStudio also comes with a Linux terminal built-in, where you can execute linux commands.
Access your Linux terminal by logging into RStudio via the web browser
(can be found in the the Getting Started section.
Here is what the RStudio dashboard looks like:
See One, Do One, Repeat: In each step of this analysis, you will first see the analysis performed with Example Sample (mini_SL342389). You will be asked to reproduce the step with that same sample and confirm you get the same results. You will then be asked to execute the step with your own assigned sample, which is found in the Getting Started section.
Do not get hung up on coding syntax: our goal is not to teach you a programming language or languages from scratch. Our goal is for you to understand the inputs, parameters, outputs, and how to interpret the output. Some participants find it helpful to think of each step as a mathematical function (e.g. y = mx + b): regardless of how complicated a block of code or command is, you are always putting data in and getting data out.
It should look like this once you're finished setting up your Linux environment:
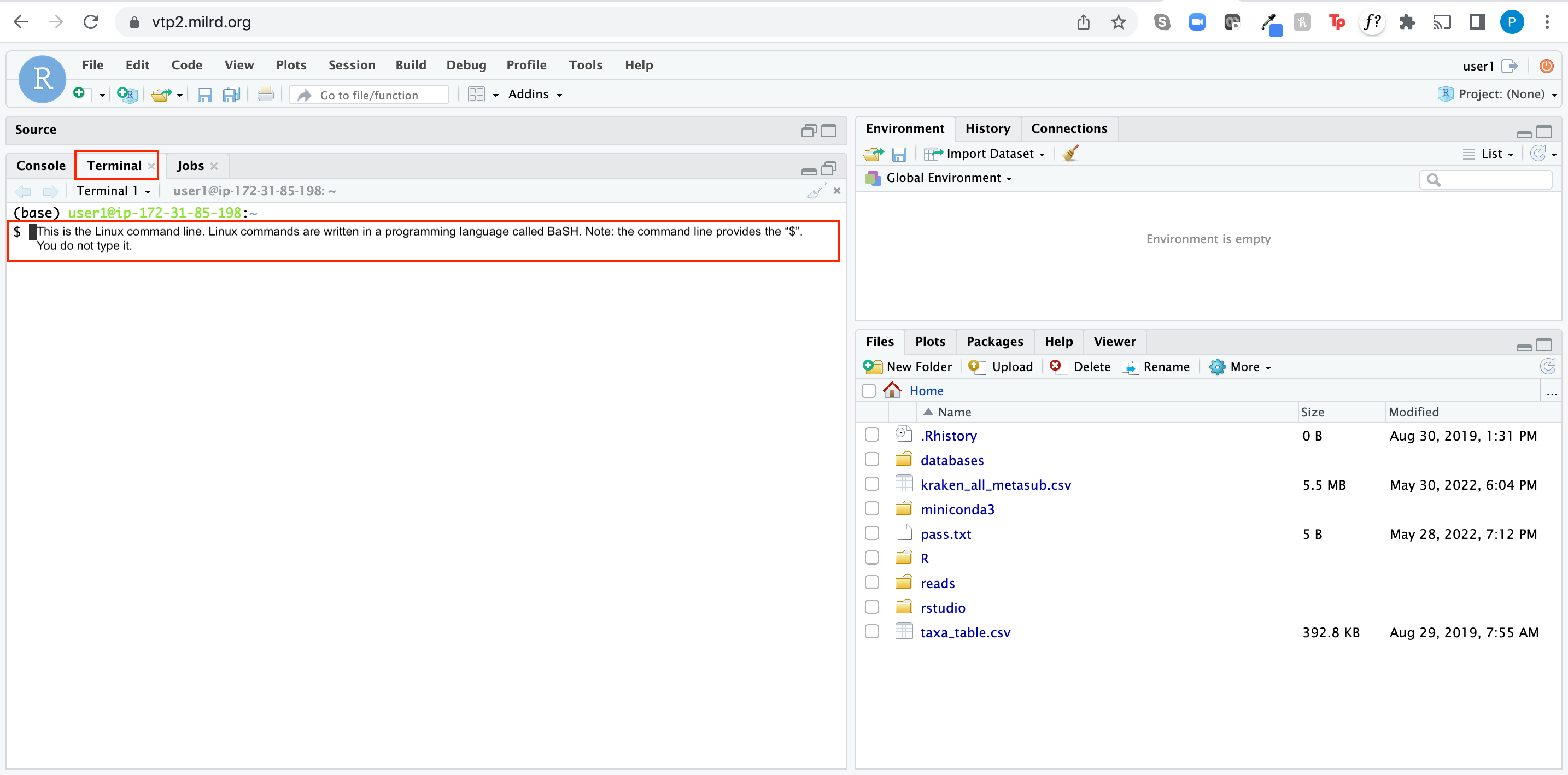
Linux is a command-line based operating system. For our purposes Linux = Unix.
The first few steps of this projects are performed in Linux. Linux is a powerful operating system used by many scientists and engineers to analyze data. Linux commands are written on the command line, which means that you tell the computer what you want it to do by typing, not by pointing and clicking like you do in operating systems like Windows, Chrome, iOS, or macOS. The commands themselves are written in a language called BaSH.
Here's a brief tutorial that covers some concepts and common commands using a sample code.
Concepts
Command Line
The Linux command line is a way of interacting with the computer's operating system by typing in commands in a text-based interface. It's a bit like talking to your computer using a special language, and that language is called Bash.
The command line in the Terminal tab should look something like this:
(base) your-user#@your-AWS-IP-address:~
$
(base) your-user#@your-AWS-IP-address:~
$ linux-commands-are-typed here
(base) your-user#@your-AWS-IP-address:~ $
Using the command line is different than using a graphical user interface (GUI) because instead of clicking on icons and buttons to interact with your computer, you type in text commands. It can take some time to learn the commands and syntax of the command line, but once you do, you can often do things faster and more efficiently than with a GUI. Some people also prefer using the command line because it gives them more control over their computer's settings and can be more powerful for certain tasks.
Let's type a command in the command line.
List the files and directories in the current directory using the ls command:
ls
You should see something like this output to the screen:
R mini_SL335868_taxa.csv mini_SL336556_taxa.csv mini_SL342390_taxa.csv miniconda3
databases mini_SL335869_taxa.csv mini_SL336557_taxa.csv mini_SL345999_taxa.csv pass.txt
kraken_all_metasub.csv mini_SL335870_taxa.csv mini_SL336558_taxa.csv mini_SL346000_taxa.csv reads
mini_SL254773_taxa.csv mini_SL335871_taxa.csv mini_SL336559_taxa.csv mini_SL346001_taxa.csv rstudio
mini_SL254775_taxa.csv mini_SL335872_taxa.csv mini_SL336560_taxa.csv mini_SL346002_taxa.csv taxa_table.csv
mini_SL254810_taxa.csv mini_SL335873_taxa.csv mini_SL336561_taxa.csv mini_SL346003_taxa.csv
mini_SL254820_taxa.csv mini_SL335875_taxa.csv mini_SL336562_taxa.csv mini_SL346004_taxa.csv
mini_SL324391_taxa.csv mini_SL336555_taxa.csv mini_SL342389_taxa.csv mini_SL346005_taxa.csv
It looks like a lot, but don't fret, this is just a bunch of files and directories (i.e. folders). You'll notice the Terminal colors by their type. For example, directories are colored blue. A directory is computer-science speak for a folder. So this tells you what folder you are currently sitting in.
Directory
As we have mentioned, a directory is another name for a folder in which files are stored.
If you look immediately to the left of the $, you will see what is called the "working directory". The ~ symbol has a special meaning in Linux systems: it refers to the home directory on your system.
Navigate to the reads directory using the cd command.
cd ~/reads/
After you execute the command, your command line should look like this:
(base) user2@ip-172-31-25-174:~/reads
$
Now, use the ls command to list the files and directories in this directory:
ls
You should see many files, most of which end in the extension fastq.gz:
SL254773.R1.fastq.gz SL335875.R1.fastq.gz SL342390.R1.fastq.gz mini_SL254775.R1.fastq.gz mini_SL336555.R1.fastq.gz mini_SL342391.R1.fastq.gz
SL254775.R1.fastq.gz SL336555.R1.fastq.gz SL342391.R1.fastq.gz mini_SL254810.R1.fastq.gz mini_SL336556.R1.fastq.gz mini_SL345999.R1.fastq.gz
SL254810.R1.fastq.gz SL336556.R1.fastq.gz SL345999.R1.fastq.gz mini_SL254820.R1.fastq.gz mini_SL336557.R1.fastq.gz mini_SL346000.R1.fastq.gz
SL254820.R1.fastq.gz SL336557.R1.fastq.gz SL346000.R1.fastq.gz mini_SL335868.R1.fastq.gz mini_SL336558.R1.fastq.gz mini_SL346001.R1.fastq.gz
SL335868.R1.fastq.gz SL336558.R1.fastq.gz SL346001.R1.fastq.gz mini_SL335869.R1.fastq.gz mini_SL336559.R1.fastq.gz mini_SL346002.R1.fastq.gz
SL335869.R1.fastq.gz SL336559.R1.fastq.gz SL346002.R1.fastq.gz mini_SL335870.R1.fastq.gz mini_SL336560.R1.fastq.gz mini_SL346003.R1.fastq.gz
SL335870.R1.fastq.gz SL336560.R1.fastq.gz SL346003.R1.fastq.gz mini_SL335871.R1.fastq.gz mini_SL336561.R1.fastq.gz mini_SL346004.R1.fastq.gz
SL335871.R1.fastq.gz SL336561.R1.fastq.gz SL346004.R1.fastq.gz mini_SL335872.R1.fastq.gz mini_SL336562.R1.fastq.gz mini_SL346005.R1.fastq.gz
SL335872.R1.fastq.gz SL336562.R1.fastq.gz SL346005.R1.fastq.gz mini_SL335873.R1.fastq.gz mini_SL342389.R1.fastq.gz
SL335873.R1.fastq.gz SL342389.R1.fastq.gz mini_SL254773.R1.fastq.gz mini_SL335875.R1.fastq.gz mini_SL342390.R1.fastq.gz
...
Now, let's say you need to go back to the home ~ directory:
cd ~
You should once again be back in the home ~ directory. It should look like this:
(base) your-user#@your-AWS-IP-address:~
$
Now, go back to the reads directory.
cd ~/reads/
Sometime we need to create a new directory to store files around a project, like when you create a new folder on your computer.
Let's create a new directory called test_directory in the current directory using the mkdir command.
mkdir test_directory
Execute ls to confirm that the test_directory directory was indeed created.
Enter the test_directory directory.
cd test_directory
Your terminal should look like this:
(base) user2@ip-172-31-25-174:~/reads/test_directory
$
Return to the ~/reads directory:
cd ~/reads
List the last last 15 commands run in your terminal so you can submit it in the next checkpoint:
history 15
There's a lot more that we could cover about working with the Linux command line, but this is enough to get started with your bioinformatics analysis.
Please take a paste your history 15 output in the Google Sheet.
In this training project, you will execute each step with an Example Sample mini_SL342389 and then your own assigned sample (Found in the Google Sheet).
To ensure each step doesn't take too long, we've taken 50,000 reads
from each assigned sample. They are labeled
with the prefix mini_ and located in the reads directory (files
without the prefix mini are the original, full-size files, which each
contain millions of reads.)
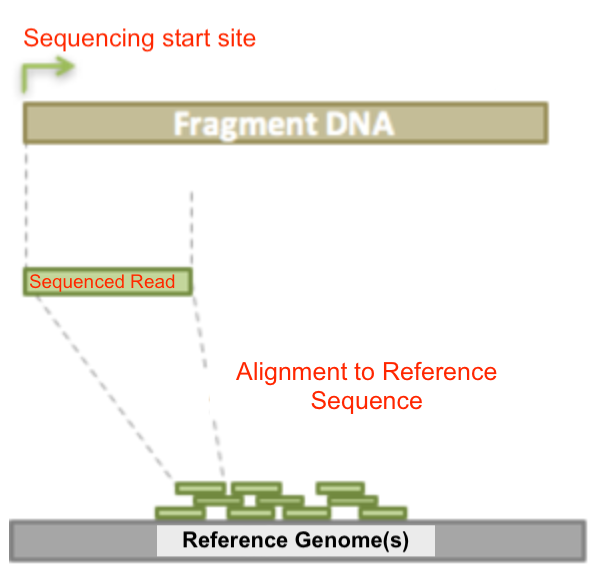
Here is an diagram that illustrates how genomic sequencing data (i.e. reads) is generated:
The raw genomic data for this project is in fastq formatted files. Fastq files have the extension .fastq.
A fastq file is a collection of fastq records.
Here's an example of a Fastq record:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
A FASTQ record has the following format:
Here's an example of a FASTQ file with two records:
@071112_SLXA-EAS1_s_7:5:1:817:345
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
@071112_SLXA-EAS1_s_7:5:1:801:338
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI
First, we have a universal step that isn't specific to any particular sample. We change to the "reads" directory by typing:
cd ~/reads/
These instructions are written assuming all Linux steps take place from the reads directory.
mini_SL342389We're going to use the head and zcat commands to look at the first few lines of our reads files.
To see the first few lines of the read 1 file, we use the zcat and head commands:
zcat mini_SL342389.R1.fastq.gz | head
$ zcat mini_SL342389.R1.fastq.gz | head
@E00438:418:HMGW3CCXY:2:1101:20547:1467 1:N:0:NAGTACGC
GGTTGCGGAGCCGGGCGAGGTAGCGGGCGCGGATCGCGGGGGCGGGGGGCAGGTCGCCCGTGGTTTCGCCGAGCCAGGGGTTGGCGAAGGTTGCGCCTTGCAGGCCGGCTTGCATCCCAAAAAGGGCGAGCG
+
AAAFJFJJFJJ7FJJFJJJJJJJJJJJJFFJJJJAJJJJJJJAFJJJJJJFFJ<AJAJJJJJJJJJJJJJJFJ7A<JJJJJJJJFJJJJJ7JJFJF<AJJAFJAAJFJFJJJ<-AFJFFJJJFFFF7FFJFF
@E00438:418:HMGW3CCXY:2:1101:14387:1309 1:N:0:NAGTACGC
CGCGCGTGTACACGCTGAAGTCCTGCCAGCGGTGGCTGCGGCTGAAGCTGGCCTGCCAGGTCCGGCTGAGCACGGGCTCGCGGCGCGGCTCCAGCGGACCGGGCGGCAGCAAGGGATCCAGCCCGAACAACGGGTCCTGCTCGAATTCAG
+
AAAFJJJJJJJJJJ<JJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJ<FJJJFJJJJFJJJJJJJJFJJJJFFJJJJJJJJJJJJJJFJJJJFJJJJJ<JJJJJJJJJJJ<J<JFJJFJJFFFAJAFF<<FJJFAJJJ<AJF7FA
@E00438:418:HMGW3CCXY:2:1101:13595:1766 1:N:0:NAGTACGC
GTCATTGTCAAACCCTCGGAAGAAACACCACTCACAACGACCTTGTTAGGTGAAGTTATGAATGAGGCAGG
The and "zcat" helps us read compressed files (the ".gz" extension means the file is compressed). The vertical bar "|" is a "pipe" that sends the output of the "zcat" command to the "head" command, which shows the first few lines of the output. If the file wasn't compressed you could simply type head name-of-file and it would output the first 10 lines to the screen.
This command "zcat" is like "cat", which shows the contents of a file, but it can read compressed files (the ".gz" extension means the file is compressed). The vertical bar "|" is a "pipe" that sends the output of the "zcat" command to the "head" command, which shows the first few lines of the output. You can still execute head name-of-compressed-file, but it will output the first 10 lines of the compressed file, witch will look like a bunch of gobbledy-gook (i.e. not human-iterpretable).
Now that you've looked at the example, let's view the first few lines of your read file for our own sample (which we're calling "mini_your_reads").
Please note: mini_your_reads is a placeholder. You need yo replace this text with name of your assigned sample:)
Look at the first few lines of your sample's read 1 file:
zcat mini_your_reads.R1.fastq.gz | head
Takeaways:
Data scientists like bioinformaticians, computer scientists, and folks in related professions will often use commands like head and zcat to look at a few lines of a file to understand it's structure or check to make sure it looks like it's supposed to.
You might ask: why not open the whole file? Two reasons: (1) files in data science are often too large to open (they can be 10s, 100s or even 1000s of gigabytes); (2) even if it's possible to open a file, it's often easier to look at the first few lines if your goal is to check or understand format.
It's important to understand the structure/format of the data you are analyzing.
In the Google Sheet, please:
Upload screenshot of your zcat results from the Example Sample
Upload screenshot of your zcat results from Your Sample
How are fastq files are structured? (i.e. What does line 1, line 2, line 3, and line 4, of each read signify?)
In this step, we're going to use a tool called Kraken to identify and quanity the microrganmisms (bacteria, archaea, and viruses in our case) based on the sequences of the dna fragments in our sample. Kraken is a software tool used in metagenomics to identify and classify the microorganisms present in a mixed DNA sample.
It is like a detective that helps scientists figure out which living things are in a sample, such as a swab from a subway, dirt, water, or even poop. It looks at tiny bits of DNA from the sample and compares them to a big library of DNA from lots of different living things, like bacteria, viruses, and fungi. Kraken then tells scientists which living things are most likely to be in the sample based on the similarities between the DNA in the sample and the DNA in the library.
Kraken uses a method called "k-mer" analysis to identify and quantify species.
To explain this in simple terms, imagine you have a long sentence made up of lots of words. A k-mer analysis would look at all the short "words" or "letter sequences" of a certain length (let's say 5 letters) in that sentence. Each unique sequence of 5 letters is called a "k-mer".
In the case of Kraken, it looks at the DNA sequences of all the organisms in a sample (which might be from a soil sample, for example) and it breaks them down into lots of short k-mers. It then compares these k-mers to a database of known k-mers from lots of different organisms.
If a particular k-mer from the sample matches a k-mer from a known organism in the database, then Kraken can say that it's likely that the organism is present in the sample. By looking at all the k-mers in the sample and comparing them to the database, Kraken can identify which organisms are likely to be present in the sample and how abundant they are.
So essentially, Kraken is like a detective that looks for clues (in the form of k-mers) to figure out which organisms are present in a sample and how many of them there are.
This is a universal step, you only need to run it one time to install the software.
Install kraken software.
conda install --channel bioconda --yes kraken
Confirm kraken was sucessfully installed.
kraken --help
This command displays the help menu for the Kraken software tool, which explains how to use the tool and what options it offers. Don't worry about reading/understanding this. We're just trying using this command to confirm the software was installed: if it wasn't you'd see an error message, not information about how to use the tool.
mini_SL342389kraken --gzip-compressed --fastq-input --threads 2 --preload --db ~/databases/minikraken_20171101_8GB_dustmasked mini_SL342389.R1.fastq.gz > mini_SL342389_taxonomic_report.csv
The output should look like this:
Loading database... complete.
50000 sequences (6.61 Mbp) processed in 44.426s (67.5 Kseq/m, 8.93 Mbp/m).
7537 sequences classified (15.07%)
42463 sequences unclassified (84.93%)
This command runs the Kraken tool on the sequencing data for Example Sample mini_SL342389.
--gzip-compressed specifies that the input files are compressed in the gzip format.--fastq-input specifies that the input files are in the FASTQ format.--threads 2 specifies that the tool should use 2 threads to speed up the analysis.--preload loads the database into memory to speed up the analysis.--db ~/databases/minikraken_20171101_8GB_dustmasked specifies the Kraken database to be used for the analysis.mini_SL342389.R1.fastq.gz is the input files containing the reads.> mini_SL342389_taxonomic_report.csv redirects the output of the tool to a CSV file named mini_SL342389_taxonomic_report.csv.Look at the the output of the kraken command.
head mini_SL342389_taxonomic_report.csv
The output to your terminal should look like this:
U E00438:418:HMGW3CCXY:2:1101:20547:1467 0 132 0:102
C E00438:418:HMGW3CCXY:2:1101:14387:1309 1163399 150 0:21 1163399:1 0:6 1163399:1 0:26 1163399:1 0:4 1163399:1 0:12 1163399:1 0:46
U E00438:418:HMGW3CCXY:2:1101:13595:1766 0 71 0:41
U E00438:418:HMGW3CCXY:2:1101:15585:1309 0 150 0:120
U E00438:418:HMGW3CCXY:2:1101:22323:1309 0 127 0:97
U E00438:418:HMGW3CCXY:2:1101:16467:1327 0 150 0:120
U E00438:418:HMGW3CCXY:2:1101:16518:1344 0 124 0:94
U E00438:418:HMGW3CCXY:2:1101:16386:1362 0 140 0:110
U E00438:418:HMGW3CCXY:2:1101:19015:1836 0 117 0:87
U E00438:418:HMGW3CCXY:2:1101:19390:1362 0 150 0:120
There are four classifications: unclassified, classified, error, or ambiguous. The output shows an identifier for each sequence, the length of the sequence, and the path through the taxonomic tree that Kraken used to classify the sequence.
Each row represents a different DNA sequence that was analyzed.
The first column shows the classification of the sequence, which can be one of four things:
U: The sequence was unclassified, meaning it did not match anything in the Kraken database C: The sequence was classified, meaning it matched a specific taxonomic group in the Kraken database E: The sequence was an error, meaning there was some problem with the analysis ?: The sequence was ambiguous, meaning it matched multiple taxonomic groups in the Kraken database
The second column shows an identifier for the sequence, which is from row 1 of a fastq read record.
The remaining columns show the path through the taxonomic tree that Kraken used to classify the sequence. For each level of the tree, there is a pair of numbers separated by a colon. The first number is the ID of a taxonomic group in the database, and the second number is the number of bases in the sequence that were classified as belonging to that group. For example, "0:117" means that 117 base pairs in the sequence were classified as belonging to the root of the taxonomic tree (i.e. they did not match any specific taxonomic group), and "A:31" means that 31 base pairs were classified as belonging to the taxonomic group identified by the ID "A".
Overall, this output tells us how many sequences were classified, how long they were, and how Kraken classified them based on their similarity to sequences in the Kraken database.
But this isn't very reader friendly, so we're gonna convert this into format called metaphlan format (denoted by a .mpa file extension) which is more human-interpretable using a tool called kraken-mpa-report
kraken-mpa-report --db ~/databases/minikraken_20171101_8GB_dustmasked mini_SL342389_taxonomic_report.csv > mini_SL342389_final_taxonomic_results.mpa
kraken-mpa-report is a command-line tool that generates a report summarizing the results of a Kraken analysis in metaphlan (.mpa) format.--db ~/databases/minikraken_20171101_8GB_dustmasked specifies the path to the Kraken database to use for the analysis. In this case, it is using a database called minikraken_20171101_8GB_dustmasked.> redirects the output of the command to a file called mini_SL342389_final_taxonomic_results.mpa. This means that the output will be saved in a file instead of being displayed on the screen.Prints the first 20 lines of the mini_SL342389_final_taxonomic_results.mpa output file to the terminal screen.
head -20 mini_SL342389_final_taxonomic_results.mpa
You should get this:
d__Viruses 20
d__Bacteria 7500
d__Archaea 7
d__Bacteria|p__Proteobacteria 3280
d__Bacteria|p__Fusobacteria 3
d__Bacteria|p__Acidobacteria 2
d__Bacteria|p__Verrucomicrobia 2
d__Bacteria|p__Kiritimatiellaeota 1
d__Bacteria|p__Planctomycetes 6
d__Bacteria|p__Bacteroidetes 126
d__Bacteria|p__Firmicutes 638
d__Bacteria|p__Deinococcus-Thermus 182
d__Bacteria|p__Actinobacteria 3151
d__Bacteria|p__Tenericutes 1
d__Archaea|p__Euryarchaeota 7
d__Bacteria|p__Proteobacteria|c__Gammaproteobacteria 2121
d__Bacteria|p__Proteobacteria|c__Alphaproteobacteria 582
d__Bacteria|p__Proteobacteria|c__Betaproteobacteria 439
d__Bacteria|p__Proteobacteria|c__Deltaproteobacteria 13
d__Bacteria|p__Proteobacteria|c__Epsilonproteobacteria 3
Print the last 20 lines of mini_SL342389_final_taxonomic_results.mpa to the terminal screen.
tail -20 mini_SL342389_final_taxonomic_results.mpa
Confirm you get this output:
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Kineosporiales|f__Kineosporiaceae|g__Kineococcus|s__Kineococcus_radiotolerans 3
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Jiangellales|f__Jiangellaceae|g__Jiangella|s__Jiangella_alkaliphila 1
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Jiangellales|f__Jiangellaceae|g__Jiangella|s__Jiangella_sp_DSM_45060 3
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Geodermatophilales|f__Geodermatophilaceae|g__Geodermatophilus|s__Geodermatophilus_obscurus 17
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Geodermatophilales|f__Geodermatophilaceae|g__Blastococcus|s__Blastococcus_saxobsidens 22
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Geodermatophilales|f__Geodermatophilaceae|g__Modestobacter|s__Modestobacter_marinus 48
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Nakamurellales|f__Nakamurellaceae|g__Nakamurella|s__Nakamurella_multipartita 9
d__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Nakamurellales|f__Nakamurellaceae|g__Nakamurella|s__Nakamurella_panacisegetis 3
d__Bacteria|p__Actinobacteria|c__Actinobacteria|g__Thermobispora|s__Thermobispora_bispora 1
d__Bacteria|p__Actinobacteria|c__Acidimicrobiia|o__Acidimicrobiales|f__Acidimicrobiaceae|g__Acidimicrobium|s__Acidimicrobium_ferrooxidans 1
d__Bacteria|p__Actinobacteria|c__Rubrobacteria|o__Rubrobacterales|f__Rubrobacteraceae|g__Rubrobacter|s__Rubrobacter_xylanophilus 1
d__Bacteria|p__Actinobacteria|c__Coriobacteriia|o__Eggerthellales|f__Eggerthellaceae|g__Eggerthella|s__Eggerthella_lenta 1
d__Bacteria|p__Actinobacteria|c__Coriobacteriia|o__Eggerthellales|f__Eggerthellaceae|g__Gordonibacter|s__Gordonibacter_urolithinfaciens 1
d__Bacteria|p__Actinobacteria|c__Thermoleophilia|o__Solirubrobacterales|f__Conexibacteraceae|g__Conexibacter|s__Conexibacter_woesei 2
d__Bacteria|p__Tenericutes|c__Mollicutes|o__Mycoplasmatales|f__Mycoplasmataceae|g__Mycoplasma|s__Mycoplasma_cynos 1
d__Archaea|p__Euryarchaeota|c__Halobacteria|o__Halobacteriales|f__Haloarculaceae|g__Halomicrobium|s__Halomicrobium_mukohataei 1
d__Archaea|p__Euryarchaeota|c__Halobacteria|o__Haloferacales|f__Haloferacaceae|g__Haloferax|s__Haloferax_volcanii 1
d__Archaea|p__Euryarchaeota|c__Halobacteria|o__Haloferacales|f__Haloferacaceae|g__Haloferax|s__Haloferax_mediterranei 1
d__Archaea|p__Euryarchaeota|c__Halobacteria|o__Natrialbales|f__Natrialbaceae|g__Haloterrigena|s__Haloterrigena_turkmenica 1
d__Archaea|p__Euryarchaeota|c__Halobacteria|o__Natrialbales|f__Natrialbaceae|g__Haloterrigena|s__Haloterrigena_daqingensis 1
Notice how these taxa are reported down to the species ("s__") level.
Run Kraken
kraken --gzip-compressed --fastq-input --threads 2 --preload --db ~/databases/minikraken_20171101_8GB_dustmasked mini_your_reads.R1.fastq.gz > mini_your_reads_taxonomic_report.csv
Look at the first 10 lines of your kraken output to ensure it looks correct.
head mini_your_reads_taxonomic_report.csv
Convert your kraken output to .mpa format.
kraken-mpa-report --db ~/databases/minikraken_20171101_8GB_dustmasked mini_your_reads_taxonomic_report.csv > mini_your_reads_final_taxonomic_results.mpa
Look at the firat 20 mines of .mpa file.
head -20 mini_your_reads_final_taxonomic_results.mpa
tail -20 mini_your_reads_final_taxonomic_results.mpa
Pull out the species level assignments in your .mpa file and store them in a .tsv file:
mini_SL342389Use the grep command to search for species-level taxonomic assignments from kraken and put them in a new file.
grep 's__' mini_SL342389_final_taxonomic_results.mpa > mini_SL342389_species_only.tsv
grep: The command-line utility used to search for specified patterns within a file.'s__': The pattern being searched for within the file.mini_SL342389_final_taxonomic_results.mpa: The input file being searched.mini_SL342389_species_only.tsv: The output file where the results of the search are being saved.Use the ls comand to confirm the mini_SL342389_species_only.tsv file was created.
Use the head utility to display the first few lines of the newly created mini_SL342389_species_only.tsv file:
head mini_SL342389_species_only.tsv
Confirm your output looks like this:
d__Viruses|f__Poxviridae|s__BeAn_58058_virus 6
d__Viruses|f__Baculoviridae|g__Alphabaculovirus|s__Lymantria_dispar_multiple_nucleopolyhedrovirus 2
d__Viruses|o__Caudovirales|f__Myoviridae|g__Kp15virus|s__Klebsiella_virus_KP15 1
d__Viruses|o__Caudovirales|f__Myoviridae|g__Secunda5virus|s__Aeromonas_virus_Aes508 1
d__Viruses|o__Caudovirales|f__Siphoviridae|s__Lactococcus_phage_WRP3 1
d__Viruses|o__Caudovirales|f__Siphoviridae|g__Pa6virus|s__Propionibacterium_virus_P104A 1
d__Viruses|s__Apis_mellifera_filamentous_virus 1
d__Viruses|o__Bunyavirales|f__Tospoviridae|g__Orthotospovirus|s__Pepper_chlorotic_spot_virus 1
d__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Thiotrichales|f__Piscirickettsiaceae|g__Methylophaga|s__Methylophaga_nitratireducenticrescens 1
d__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Pseudomonadales|f__Moraxellaceae|g__Acinetobacter|s__Acinetobacter_lwoffii 60
You want to confirn that each line is taxonomically classified down to the speciess level by confirming an s__ is listed.
Count the number of lines in the mini_SL342389_species_only.tsv file.
wc -l mini_SL342389_species_only.tsv
915 mini_SL342389_species_only.tsv
This result indicates you could 987 lines in your mini_SL342389_species_only.tsv file, which means you identified 987 species in Example Sample mini_SL342389 (since each line is one species).
The rationale behind executing these commands is to extract and isolate the taxonomic assignments at the species level from the original file and to obtain a count of the number of species identified in the sample. The grep command filters the original file to only include lines with species-level assignments, and the head command is used to quickly check the output file to ensure that the command has worked as intended. Finally, the wc command is used to obtain a count of the number of lines (i.e., species) in the output file.
grep 's__' mini_your_reads_final_taxonomic_results.mpa > mini_your_reads_species_only.tsv
head mini_your_reads_species_only.tsv
wc -l mini_your_reads_species_only.tsv
In the Google Sheet, please:
Upload a screenshot of your kraken command output for the Example Sample. (i.e. the one that states the % sequences classified and unclassified)
Upload a screenshot of your kraken command output for Your Sample.
Upload a screenshot of your head -20 and tail -20 results for the Example Sample.
Upload a screenshot of your head -20 and tail -20 results for Your Sample.
Answer these questions:
a. What do d, p, and other abbreviations stand for? b. What do you the numbers at the end of each line signify? c. Why do you think Kraken can't characterize some reads at all? d. Why do you think Kraken can't characterize some reads down to the species level? e. When identifying only the species-level taxonomic assignments, what "search term" do you use in your code?
For the next set of steps, we're going to use the R programming language.
Please make sure the RStudio user interface dashboard looks as follows: Console Pane (Lower Left Quadrant), Scripting Pane (Upper Left Quadrant), File Manager/Plots Display/Help-Tab Pane (Lower Right Quadrant), Environment/History-Tab Pane (Upper Right Quadrant).
In R, code is always executed via the Console, but you have the choice whether to execute that code in a Script (opened in the Scripting Pane) or directly in the Console.
Unless you need to quicly execute a
one-liner (e.g. setting a working directory using the setwd command),
you'll want to be writing your code in a script, highlighting it, and
clicking Run to execute it in the console. This is because you can
easily edit code in a script and re-run it. Once code is run in the
Console it is not editable.
In R, code is always executed via the Console, but you have the choice whether to execute that code in a Script (opened in the Scripting Pane) or directly in the Console. Unless you need to quicly execute a one-liner (e.g. setting a working directory using the setwd command), you'll want to be writing your code in a script, highlighting it, and clicking Run to execute it in the console. This is because you can easily edit code in a script and re-run it. Code run in the Console it is not editable in the Console.
Let's complete an R Tutorial, partly based on this website.
Instructions First execute the codes in the Console. Then clear your Console Pane and Environment Panes using the "brush" icons, re-enter the same codes in a script in the Scripting Pane (there is an empty script created for you called "Untitled1"), highlight the codes and click "Run". The results should be the same as when executed in the Console, but you can now edit the script and re-run them as much as you'd like.
R Session After R is started, there is a console awaiting for input. At the prompt (>), you can enter numbers and perform calculations.
Add two numbers:
1 + 2
You should get:
[1] 3
Variable Assignment We assign values to variables with the assignment operator "=". Just typing the variable by itself at the prompt will print out the value. We should note that another form of assignment operator "<-" is also in use.
Let's try assigning a variable:
x = 1
x you should see it print the value you assigned it. Let's try:
x
You should see:
[1] 1
Functions R functions are invoked by its name, then followed by the parenthesis, and zero or more arguments.
Let's try some common functions
mean() - calculates the arithmetic mean of a numeric vector:
vec <- c(1, 2, 3, 4, 5)
mean(vec) # should return 3
sum(vec) # should return 15
All text after the pound sign "#" within the same line is considered a comment:
1 + 1 # this is a comment
Extension Packages
Sometimes we need additional functionality beyond those offered by the core R library. In order to install an extension package, you should invoke the install.packages() function at the prompt and follow the instruction.
When you want to use that library you need to invoke it in your script using the library() function so R knows it needs to open it.
Let's try this with the package ggplot2:
library(ggplot2)
Let's make a simple plot using the ggplot() function from the ggplot2 library:
# Load ggplot2 package
library(ggplot2)
# Create a data frame with two variables
data <- data.frame(x = c(1, 2, 3, 4, 5), y = c(2, 4, 5, 4, 6))
# Create a scatter plot using ggplot2
ggplot(data, aes(x = x, y = y)) +
geom_point() # Add points to the plot
In the Google Sheet:
mini_SL342389#This line reads in the mini_SL342389_species_only.tsv file using the read.csv() function.
taxa_mini_SL342389_species = read.csv('~/reads/mini_SL342389_species_only.tsv', header=FALSE, sep='\t')
#This line orders the data in the taxa_mini_SL342389_species variable based on the values in the second column (V2) in descending order.
taxa_mini_SL342389_species_ordered <- taxa_mini_SL342389_species[order(taxa_mini_SL342389_species$V2, decreasing = TRUE), ]
#This line outputs the first five rows of the ordered data in the taxa_mini_SL342389_species_ordered variable.
head(taxa_mini_SL342389_species_ordered, 5)
The code reads in a file called mini_SL342389_species_only.tsv and stores the data in a variable called taxa_mini_SL342389_species. Then, it orders the data based on the second column (V2) in descending order and stores the new ordered data in a new variable called taxa_mini_SL342389_species_ordered. Finally, the code outputs the top 5 rows of the ordered data.
These five rows are the top 5 species in your dataset.
taxa_yourSample_species <- read.csv('~/reads/mini_your_reads_species_only.tsv', header=FALSE, sep='\t')
taxa_yourSample_species_ordered <- taxa_yourSample_species[order(taxa_yourSample_species$V2, decreasing = TRUE), ]
head(taxa_yourSample_species_ordered, 5)
In the Google Sheet:
mini_SL342389Abundance of Phyla
library(ggplot2)
taxa_mini_SL342389 <- read.csv('~/mini_SL342389_taxa.csv', header=TRUE, sep=',')
phyla_mini_SL342389 <- taxa_mini_SL342389[taxa_mini_SL342389$rank == 'phylum',]
phyla_mini_SL342389 <- phyla_mini_SL342389[phyla_mini_SL342389$percent_abundance >= 0.3,]
ggplot(phyla_mini_SL342389 , aes(x=sample, y=percent_abundance, fill=taxon)) +
geom_bar(stat="identity") +
xlab('Sample') +
ylab('Abundance') +
labs(fill='Phylum') +
theme(axis.text.x = element_text(angle = 90))
Abundance of Families
library(ggplot2)
taxa_mini_SL342389 <- read.csv('~/mini_SL342389_taxa.csv', header=TRUE, sep=',')
family_mini_SL342389 <- taxa_mini_SL342389[taxa_mini_SL342389$rank == 'family',]
family_mini_SL342389 <- family_mini_SL342389[family_mini_SL342389$percent_abundance >= 0.3,]
ggplot(family_mini_SL342389 , aes(x=sample, y=percent_abundance, fill=taxon)) +
geom_bar(stat="identity") +
xlab('Sample') +
ylab('Abundance') +
labs(fill='Family') +
theme(axis.text.x = element_text(angle = 90))
Abundance of Phyla
library(ggplot2)
taxa_yourSample <- read.csv('~/yourSample_taxa.csv', header=TRUE, sep=',')
phylum_yourSample <- taxa_yourSample[taxa_yourSample$rank == 'phylum',]
phylum_yourSample <- phylum_yourSample[family_yourSample$percent_abundance >= 0.3,]
ggplot(phylum_yourSample, aes(x=sample, y=percent_abundance, fill=taxon)) +
geom_bar(stat="identity") +
xlab('Sample') +
ylab('Abundance') +
labs(fill='Phylum') +
theme(axis.text.x = element_text(angle = 90))
Abundance of Families
library(ggplot2)
taxa_yourSample <- read.csv('~/yourSample_taxa.csv', header=TRUE, sep=',')
family_yourSample <- taxa_yourSample[taxa_yourSample$rank == 'family',]
family_yourSample <- family_yourSample[family_yourSample$percent_abundance >= 0.3,]
ggplot(family_yourSample, aes(x=sample, y=percent_abundance, fill=taxon)) +
geom_bar(stat="identity") +
xlab('Sample') +
ylab('Abundance') +
labs(fill='Family') +
theme(axis.text.x = element_text(angle = 90))
Barplot: Abundance of top phyla across all samples
library(ggplot2)
taxa_all_barplot <- read.csv('~/kraken_all_metasub.csv', header=TRUE, sep=',')
taxa_all_barplot <- taxa_all_barplot[taxa_all_barplot$rank == 'phylum',]
taxa_all_barplot <- taxa_all_barplot[taxa_all_barplot$percent_abundance >= 0.3,]
ggplot(taxa_all_barplot, aes(x=sample, y=percent_abundance, fill=taxon)) +
geom_bar(stat="identity") +
xlab('Sample') +
ylab('Abundance') +
labs(fill='Phylum') +
theme(axis.text.x = element_text(angle = 90))
In the Google Sheet:
Identify resistance genes in your sample’s most abundant species
Navigate to the Bacterial and Viral Bioinformatics Resource Center (BV-BRC)
Use the tools in BV-BRC to ID at least one AMR locus/gene for the top 5 most abundant species in your sample.
Here’s a screencast using the species Staphylococcus aureus (Methicillin-resistant Staphylococcus aureus (MRSA) is a widely known antibiotic resistant hospital acquired infection):
In the Google Sheet:
List three efflux pump conferring antibiotic resistance locus IDs for the species you queried using the BV-BRC. (Only report the Locus ID, which is denoted by a > symbol. Do not report the locus sequence.)
Explain how to deterimine if a resistant strain of one of the abundant species in your sample is present.
So, at this point, you've made it through a bioinformatic analysis of a metagenomic dataset. Well done!
At this point, we want to show you how what you did compared to what a professional computational biologist would do. Becuase we had limited time, and had so much material to get through, we simplified the analysis you completed by removing some steps:
Normally, professional computational biologists would take the raw data through a quality control process, where they review the quality scores of the nucleotides and trim poor-quality nucleotides (if it's just a few) and remove whole reads from the dataset if it constitutes a lot of the read.
In metagenomics analysis there are often DNA fragments from species we don't care about and that can complicate analysis. For example, if you collect DNA from surfaces that humans touch like is the case in transit systems, you tend to get a lot of contaminating human reads in your data. To remove this, we often use an algorithm called an aligner which tries to match each read to a reference genome of the species we want to remove. If a read matches, it gets removed so it doesn't undermine downstream analysis.
There would be many more detailed analyses a professional could, and likely would, perform downstream. For example, a scientist might want to know if a bacterial species in a sample is resistant strain.
Here's a flowchart with some of the additional steps:
Please complete the Post-VTP Survey.